Commands
The CLI performs operations in the user workspace context by default. Use the TOWER_WORKSPACE_ID environment variable or the --workspace parameter to specify an organization workspace ID.
Use the -h or --help parameter to list the available commands and their associated options.
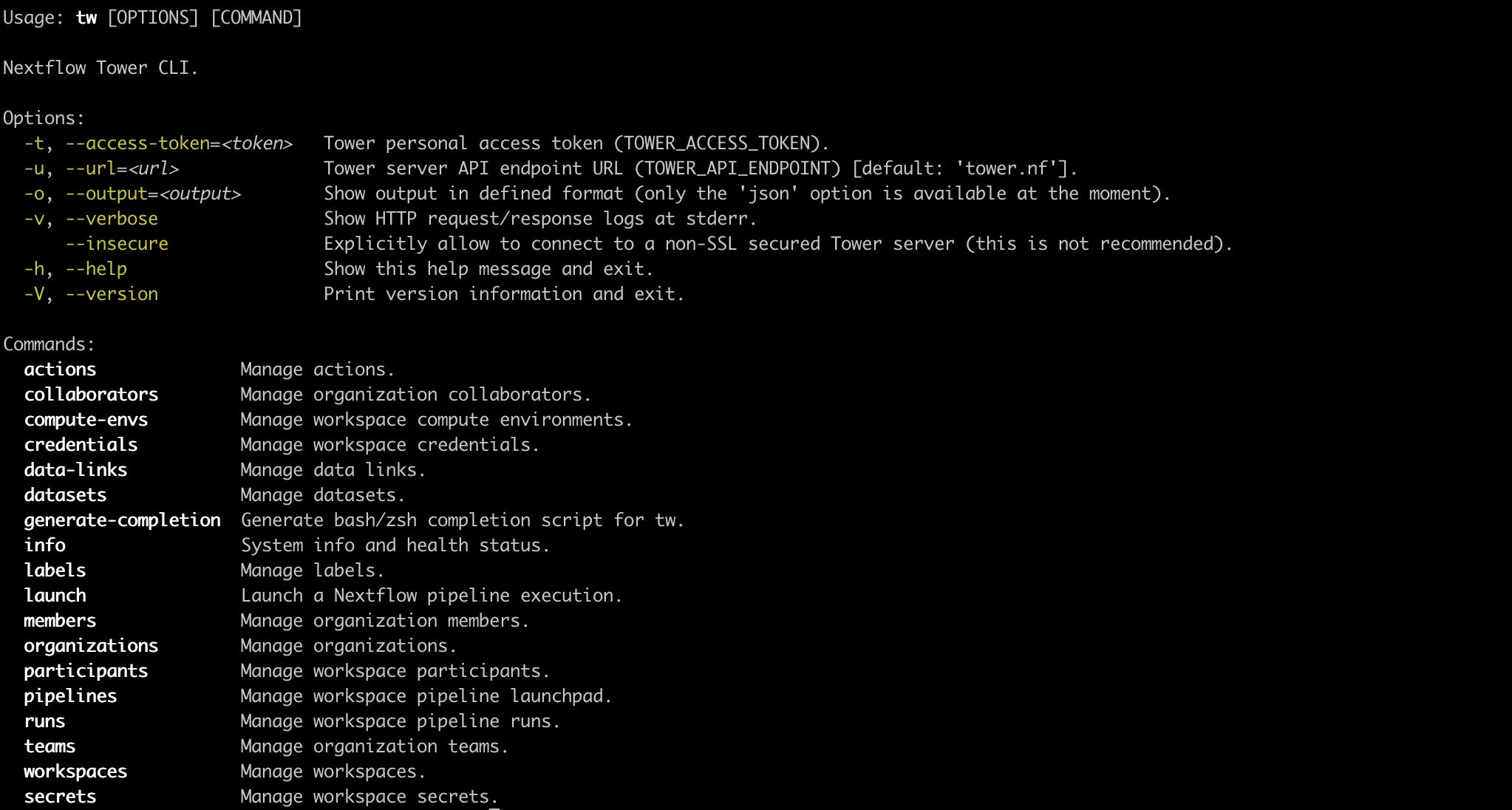
For help with a specific subcommand, run the command with -h or --help appended. For example, tw credentials add google -h.
Use tw --output=json <command> to dump and store Seqera Platform entities in JSON format.
Use tw --output=json <command> | jq -r '.[].<key>' to pipe the command to use jq to retrieve specific values in the JSON output. For example, tw --output=json workspaces list | jq -r '.workspaces[].orgId' returns the organization ID for each workspace listed.
Credentials
To launch pipelines in a Platform workspace, you need credentials for:
- Compute environments
- Pipeline repository Git providers
- (Optional) Tower agent — used with HPC clusters
- (Optional) Container registries, such as docker.io
Add credentials
Run tw credentials add -h to view a list of providers.
Run tw credentials add <provider> -h to view the required fields for your provider.
You can add multiple credentials from the same provider in the same workspace.
Compute environment credentials
Platform requires credentials to access your cloud compute environments. See the compute environment page for your cloud provider for more information.
tw credentials add aws --name=my_aws_creds --access-key=<aws access key> --secret-key=<aws secret key>
New AWS credentials 'my_aws_creds (1sxCxvxfx8xnxdxGxQxqxH)' added at user workspace
Git credentials
Platform requires access credentials to interact with pipeline Git repositories. See Git integration for more information.
tw credentials add github -n=my_GH_creds -u=<GitHub username> -p=<GitHub access token>
New GITHUB credentials 'my_GH_creds (xxxxx3prfGlpxxxvR2xxxxo7ow)' added at user workspace
Container registry credentials
Configure credentials for the Nextflow Wave container service to authenticate to private and public container registries. See the Container registry credentials section under Credentials for registry-specific instructions.
Container registry credentials are only used by the Wave container service. See Wave containers for more information.
List credentials
tw credentials list
Credentials at user workspace:
ID | Provider | Name | Last activity
------------------------+-----------+------------------------------------+-------------------------------
1x1HxFxzxNxptxlx4xO7Gx | aws | my_aws_creds_1 | Wed, 6 Apr 2022 08:40:49 GMT
1sxCxvxfx8xnxdxGxQxqxH | aws | my_aws_creds_2 | Wed, 9 Apr 2022 08:40:49 GMT
2x7xNsf2xkxxUIxXKxsTCx | ssh | my_ssh_key | Thu, 8 Jul 2021 07:09:46 GMT
4xxxIeUx7xex1xqx1xxesk | github | my_github_cred | Wed, 22 Jun 2022 09:18:05 GMT
Delete credentials
tw credentials delete --name=my_aws_creds
Credentials '1sxCxvxfx8xnxdxGxQxqxH' deleted at user workspace
Compute environments
Compute environments define the execution platform where a pipeline runs. A compute environment is composed of the credentials, configuration, and storage options related to a particular computing platform. See Compute environments for more information on supported providers.
Run tw compute-envs -h to view the list of supported compute environment operations.
Add a compute environment
Run tw compute-envs add -h to view the list of supported providers.
Run tw compute-envs add <platform> -h to view the required and optional fields for your provider.
You must add the credentials for your provider before creating your compute environment.
tw compute-envs add aws-batch forge --name=my_aws_ce \
--credentials=<my_aws_creds_1> --region=eu-west-1 --max-cpus=256 \
--work-dir=s3://<bucket name> --wait=AVAILABLE
New AWS-BATCH compute environment 'my_aws_ce' added at user workspace
This command will:
- Use Batch Forge to automatically manage the AWS Batch resource lifecycle (
forge) - Use the credentials previously added to the workspace (
--credentials) - Create the required AWS Batch resources in the AWS Ireland (
eu-west-1) region - Provision a maximum of 256 CPUs in the compute environment (
--max-cpus) - Use an existing S3 bucket to store the Nextflow work directory (
--work-dir) - Wait until the compute environment has been successfully created and is ready to use (
--wait)
See the compute environment page for your provider for detailed information on Batch Forge and manual compute environment creation.
Delete a compute environment
tw compute-envs delete --name=my_aws_ce
Compute environment '1sxCxvxfx8xnxdxGxQxqxH' deleted at user workspace
Default compute environment
Select a primary compute environment to be used by default in a workspace. You can override the workspace primary compute environment by explicitly specifying an alternative compute environment when you create or launch a pipeline.
tw compute-envs primary set --name=my_aws_ce
Primary compute environment for workspace 'user' was set to 'my_aws_ce (1sxCxvxfx8xnxdxGxQxqxH)'
Import and export a compute environment
Export the configuration details of a compute environment in JSON format for scripting and reproducibility purposes.
tw compute-envs export --name=my_aws_ce my_aws_ce_v1.json
Compute environment exported into 'my_aws_ce_v1.json'
Similarly, a compute environment can be imported to a workspace from a previously exported JSON file.
tw compute-envs import --name=my_aws_ce_v1 ./my_aws_ce_v1.json
New AWS-BATCH compute environment 'my_aws_ce_v1' added at user workspace
Datasets
Run tw datasets -h to view the list of supported operations.
Datasets are CSV (comma-separated values) and TSV (tab-separated values) files stored in a workspace, used as inputs during pipeline execution. The most commonly used datasets for Nextflow pipelines are samplesheets, where each row consists of a sample, the location of files for that sample (such as FASTQ files), and other sample details.
Add a dataset
Run tw datasets add -h to view the required and optional fields for adding a dataset.
Add a preconfigured dataset file to a workspace (include the --header flag if the first row of your samplesheet file is a header):
tw datasets add --name=samplesheet1 --header samplesheet_test.csv
Dataset 'samplesheet1' added at user workspace with id '60gGrD4I2Gk0TUpEGOj5Td'
The maximum supported dataset file size is 10 MB.
Delete a dataset
To delete a workspace dataset, specify either the dataset name (-n flag) or ID (-i flag):
tw datasets delete -i 6tYMjGqCUJy6dEXNK9y8kh
Dataset '6tYMjGqCUJy6dEXNK9y8kh' deleted at 97652229034604 workspace
Download a dataset
View a stored dataset's contents:
tw datasets download -n samplesheet1
sample,fastq_1,fastq_2,strandedness
WT_REP1,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357070_1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357070_2.fastq.gz,auto
WT_REP1,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357071_1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357071_2.fastq.gz,auto
WT_REP2,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357072_1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357072_2.fastq.gz,reverse
RAP1_UNINDUCED_REP1,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357073_1.fastq.gz,,reverse
RAP1_UNINDUCED_REP2,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357074_1.fastq.gz,,reverse
RAP1_UNINDUCED_REP2,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357075_1.fastq.gz,,reverse
RAP1_IAA_30M_REP1,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357076_1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/testdata/GSE110004/SRR6357076_2.fastq.gz,reverse
List datasets
Run tw datasets list -h to view the optional fields for listing and filtering datasets.
tw datasets list -f data
Datasets at 97652229034604 workspace:
ID | Name | Created
------------------------+----------+-------------------------------
6vBGj6aWWpBuLpGKjJDpZy | dataset2 | Tue, 27 Aug 2024 14:49:32 GMT
View a dataset
Run tw datasets view -h to view the required and optional fields for viewing a stored dataset's details.
tw datasets view -n samplesheet1
Dataset at 97652229034604 workspace:
-------------+-------------------------------
ID | 60gGrD4I2Gk0TUpEGOj5Td
Name | samplesheet1
Description |
Media Type | text/csv
Created | Mon, 19 Aug 2024 07:59:16 GMT
Updated | Mon, 19 Aug 2024 07:59:17 GMT
Update a dataset
Run tw datasets update -h to view the required and optional fields for updating a dataset.
tw datasets update -n dataset1 --new-name=dataset2 -f samplesheet_test.csv
Dataset 'dataset1' updated at 97652229034604 workspace with id '6vBGj6aWWpBuLpGKjJDpZy'
Obtain a dataset URL
Run tw datasets url -h to view the required and optional fields for obtaining dataset URLs.
tw datasets url -n dataset2
Dataset URL
-----------
https://api.cloud.seqera.io/workspaces/97652229034xxx/datasets/6vBGj6aWWpBuLpGKjJDxxx/v/2/n/samplesheet_test.csv
Pipelines
Run tw pipelines -h to view the list of supported operations.
Pipelines define pre-configured workflows in a workspace. A pipeline consists of a workflow repository, launch parameters, and a compute environment.
Add a pipeline
Run tw pipelines add -h to view the required and optional fields for adding your pipeline.
Add a pre-configured pipeline to the Launchpad:
tw pipelines add --name=my_rnaseq_nf_pipeline \
--params-file=my_rnaseq_nf_pipeline_params.yaml \
--config=<path/to/nextflow/conf/file> \
https://github.com/nextflow-io/rnaseq-nf
New pipeline 'my_rnaseq_nf_pipeline' added at user workspace
The optional --params-file flag is used to pass a set of default parameters that will be associated with the pipeline in the Launchpad.
The optional --config flag is used to pass a custom Nextflow configuration file — configuration values passed here override the same values in the default pipeline repository nextflow.conf file. When this flag is set, all configuration values specified in Platform pipeline or compute environment Nextflow config fields are ignored.
The params-file or --config file must be a YAML or JSON file using Nextflow configuration syntax.
Import and export a pipeline
Export the configuration details of a pipeline in JSON format for scripting and reproducibility purposes.
tw pipelines export --name=my_rnaseq_nf_pipeline my_rnaseq_nf_pipeline_v1.json
Pipeline exported into 'my_rnaseq_nf_pipeline_v1.json'
Similarly, a pipeline can be imported to a workspace from a previously exported JSON file.
tw pipelines import --name=my_rnaseq_nf_pipeline_v1 ./my_rnaseq_nf_pipeline_v1.json
New pipeline 'my_rnaseq_nf_pipeline_v1' added at user workspace
Update a pipeline
The default launch parameters can be changed with the update command:
tw pipelines update --name=my_rnaseq_nf_pipeline \
--params-file=my_rnaseq_nf_pipeline_params_2.yaml
Launch pipelines
Run tw launch -h to view supported launch options.
Launch a preconfigured pipeline
If no custom parameters are passed via the CLI during launch, the defaults set for the pipeline in the Launchpad will be used.
tw CLI users are bound to the same user permissions that apply in the Platform UI. Launch users can launch pre-configured pipelines in the workspaces they have access to, but they cannot add or run new pipelines.
tw launch my_rnaseq_nf_pipeline \
--config=<path/to/nextflow/conf/file> \
Workflow 1XCXxX0vCX8xhx submitted at user workspace.
https://cloud.seqera.io/user/user1/watch/1XCXxX0vCX8xhx
The optional --config flag is used to pass a custom Nextflow configuration file — configuration values passed here override the same values in the default pipeline repository nextflow.conf file. When this flag is set, all configuration values specified in Platform pipeline or compute environment Nextflow config fields are ignored.
When using --wait, tw can exit with one of two exit codes:
0: When the run reaches the desired state.1: When the run reaches a state that makes it impossible to reach the desired state.
Use --wait=SUCCEEDED if you want the command to wait until the pipeline execution is complete.
Launch a pipeline with custom parameters
To specify custom parameters during pipeline launch, specify a custom --params-file:
tw launch my_rnaseq_nf_pipeline --params-file=my_rnaseq_nf_pipeline_params_2.yaml
Workflow 2XDXxX0vCX8xhx submitted at user workspace.
https://cloud.seqera.io/user/user1/watch/2XDXxX0vCX8xhx
See Nextflow configuration for more information.
Launch an unsaved pipeline
The CLI can directly launch pipelines that have not been added to the Launchpad in a Platform workspace by using the full pipeline repository URL:
tw launch https://github.com/nf-core/rnaseq \
--params-file=./custom_rnaseq_params.yaml \
--config=<path/to/nextflow/conf/file> \
--compute-env=my_aws_ce --revision 3.8.1 \
--profile=test,docker
Workflow 2XDXxX0vCX8xhx submitted at user workspace.
https://cloud.seqera.io/user/user1/watch/2XDXxX0vCX8xhx
- Pipeline parameters are defined within the
custom_rnaseq_params.yamlfile. - The optional
--configflag is used to pass a custom Nextflow configuration file — configuration values passed here override the same values in the default pipeline repositorynextflow.conffile. When this flag is set, all configuration values specified in Platform pipeline or compute environment Nextflow config fields are ignored. - Other parameters such as
--profileand--revisioncan also be specified. - A non-primary compute environment can be used to launch the pipeline. Omit
--compute-envto launch with the workspace default compute environment.
CLI users are bound to the same user permissions that apply in the Platform UI. Launch users can launch pre-configured pipelines in the workspaces they have access to, but they cannot add or run new pipelines.
Runs
Run tw runs -h to view supported runs operations.
Runs display all the current and previous pipeline runs in the specified workspace. Each new or resumed run is given a random name such as grave_williams by default, which can be overridden with a custom value at launch. See Run details for more information. As a run executes, it can transition through the following states:
submitted: Pending executionrunning: Runningsucceeded: Completed successfullyfailed: Successfully executed, where at least one task failed with a terminate error strategycancelled: Stopped manually during executionunknown: Indeterminate status
View pipeline's runs
Run tw runs view -h to view all the required and optional fields for viewing a pipeline's runs.
tw runs view -i 2vFUbBx63cfsBY -w seqeralabs/showcase
Run at [seqeralabs / showcase] workspace:
General
---------------------+-------------------------------------------------
ID | 2vFUbBx63cfsBY
Operation ID | b5d55384-734e-4af0-8e47-0d3abec71264
Run name | adoring_brown
Status | SUCCEEDED
Starting date | Fri, 31 May 2024 10:38:30 GMT
Commit ID | b89fac32650aacc86fcda9ee77e00612a1d77066
Session ID | 9365c6f4-6d79-4ca9-b6e1-2425f4d957fe
Username | user1
Workdir | s3://seqeralabs-showcase/scratch/2vFUbBx63cfsBY
Container | No container was reported
Executors | awsbatch
Compute Environment | seqera_aws_ireland_fusionv2_nvme
Nextflow Version | 23.10.1
Labels | star_salmon,yeast
List runs
Run tw runs list -h to view all the required and optional fields for listing runs in a workspace.
tw runs list
Pipeline runs at [seqeralabs / testing] workspace:
ID | Status | Project Name | Run Name | Username | Submit Date
----------------+-----------+----------------------------+---------------------------------+-----------------------+-------------------------------
49Gb5XVMud2e7H | FAILED | seqeralabs/nf-aggregate | distraught_archimedes | user1 | Fri, 31 May 2024 16:22:10 GMT
4anNFvTUwRFDp | SUCCEEDED | nextflow-io/rnaseq-nf | nasty_kilby | user1 | Fri, 31 May 2024 15:23:12 GMT
3wo3Kfni6Kl3hO | SUCCEEDED | nf-core/proteinfold | reverent_linnaeus | user2 | Fri, 31 May 2024 15:22:38 GMT
<snip>
4fIRrFgZV3eDb1 | FAILED | nextflow-io/hello | gigantic_lichterman | user1 | Mon, 29 Apr 2024 08:44:47 GMT
cHEdKBXmdoQQM | FAILED | mathysgrapotte/stimulus | mighty_poitras | user3 | Mon, 29 Apr 2024 08:08:52 GMT
Use the optional --filter flag to filter the list of runs returned by one or more keyword:value entries:
statuslabelworkflowIdrunNameusernameprojectNameafterbeforesessionIdis:starred
If no keyword is defined, the filtering is applied to the runName, projectName (the pipeline name), and username.
The after and before flags require an ISO 8601 timestamp with UTC timezone (YYYY-MM-DDThh:mm:ss.sssZ).
tw runs list --filter hello_slurm_20240530
Pipeline runs at [seqeralabs / showcase] workspace:
ID | Status | Project Name | Run Name | Username | Submit Date
---------------+-----------+-------------------+--------------------------------------+------------+-------------------------------
pZeJBOLtIvP7R | SUCCEEDED | nextflow-io/hello | hello_slurm_20240530_e75584566f774e7 | user1 | Thu, 30 May 2024 09:12:51 GMT
Multiple filter criteria can be defined:
tw runs list --filter="after:2024-05-29T00:00:00.000Z before:2024-05-30T00:00:00.000Z username:user1"
Pipeline runs at [seqeralabs / testing] workspace:
ID | Status | Project Name | Run Name | Username | Submit Date
----------------+-----------+-----------------------+--------------------+-------------+-------------------------------
xJvK95W6YUmEz | SUCCEEDED | nextflow-io/rnaseq-nf | ondemand2 | user1 | Wed, 29 May 2024 20:35:28 GMT
1c1ckn9a3j0xF0 | SUCCEEDED | nextflow-io/rnaseq-nf | fargate | user1 | Wed, 29 May 2024 20:28:02 GMT
3sYX1acJ01T7rL | SUCCEEDED | nextflow-io/rnaseq-nf | min1vpcu-spot | user1 | Wed, 29 May 2024 20:27:47 GMT
4ZYJGWJCttXqXq | SUCCEEDED | nextflow-io/rnaseq-nf | min1cpu-ondemand | user1 | Wed, 29 May 2024 20:25:21 GMT
4LCxsffTqf3ysT | SUCCEEDED | nextflow-io/rnaseq-nf | lonely_northcutt | user1 | Wed, 29 May 2024 20:09:51 GMT
4Y8EcyopNiYBlJ | SUCCEEDED | nextflow-io/rnaseq-nf | fargate | user1 | Wed, 29 May 2024 18:53:47 GMT
dyKevNwxK50XX | SUCCEEDED | mark814/nr-test | cheeky_cuvier | user1 | Wed, 29 May 2024 12:21:10 GMT
eS6sVB5A387aR | SUCCEEDED | mark814/nr-test | evil_murdock | user1 | Wed, 29 May 2024 12:11:08 GMT
A leading and trailing * wildcard character is supported:
tw runs list --filter="*man/rnaseq-*"
Pipeline runs at [seqeralabs / testing] workspace:
ID | Status | Project Name | Run Name | Username | Submit Date
----------------+-----------+---------------------+---------------------+----------------+-------------------------------
5z4AMshti4g0GK | SUCCEEDED | robnewman/rnaseq-nf | admiring_darwin | user1 | Tue, 16 Jan 2024 19:56:29 GMT
62LqiS4O4FatSy | SUCCEEDED | robnewman/rnaseq-nf | cheeky_yonath | user1 | Wed, 3 Jan 2024 12:36:09 GMT
3k2nu8ZmcBFSGv | SUCCEEDED | robnewman/rnaseq-nf | compassionate_jones | user3 | Tue, 2 Jan 2024 16:22:26 GMT
3zG2ggf5JsniNW | SUCCEEDED | robnewman/rnaseq-nf | fervent_payne | user1 | Wed, 20 Dec 2023 23:55:17 GMT
1SNIcSXRuJMSNZ | SUCCEEDED | robnewman/rnaseq-nf | curious_babbage | user3 | Thu, 28 Sep 2023 17:48:04 GMT
5lI2fZUZfiokBI | SUCCEEDED | robnewman/rnaseq-nf | boring_heisenberg | user2 | Thu, 28 Sep 2023 12:29:27 GMT
5I4lsRXIHVEjNB | SUCCEEDED | robnewman/rnaseq-nf | ecstatic_ptolemy | user2 | Wed, 27 Sep 2023 22:06:19 GMT
Relaunch run
Run tw runs relaunch -h to view all the required and optional fields for relaunching a run in a workspace.
Cancel a run
Run tw runs cancel -h to view all the required and optional fields for canceling a run in a workspace.
Manage labels for runs
Run tw runs labels -h to view all the required and optional fields for managing labels for runs in a workspace.
In the example below, we add the labels test and rnaseq-demo to the run with ID 5z4AMshti4g0GK:
tw runs labels -i 5z4AMshti4g0GK test,rnaseq-demo
'set' labels on 'run' with id '5z4AMshti4g0GK' at 34830707738561 workspace
Delete a run
Run tw runs delete -h to view all the required and optional fields for deleting a run in a workspace.
Dump all logs and details of a run
Run tw runs dump -h to view all the required and optional fields for dumping all logs and details of a run in a workspace. The supported formats are .tar.xz and .tar.gz. In the example below, we dump all the logs and details for the run with ID 5z4AMshti4g0GK to the output file file.tar.gz.
tw runs dump -i 5z4AMshti4g0GK -o file.tar.gz
- Tower info
- Workflow details
- Task details
Pipeline run '5z4AMshti4g0GK' at [seqeralabs / testing] workspace details dump at 'file.tar.gz'
Labels
Run tw labels -h to view supported label operations.
Manage labels and resource labels.
Add a label
Run tw labels add -h to view the required and optional fields for adding a label.
Resource labels consist of a name=value pair.
Labels require only a name.
tw labels add -n Label1 -w DocTestOrg2/Testing -v Value1
Label 'Label1=Value1' added at 'DocTestOrg2/Testing' workspace with id '268741348267491'
List labels
Run tw labels list -h to view the optional fields for filtering labels.
tw labels list
Labels at 97652229034604 workspace:
ID | Name | Value | Type
-----------------+------------------------+-----------+----------
116734717739444 | manual-fusion-amd64 | | Normal
120599302764779 | test-with-prefix | | Normal
128477232893714 | manual-fusion-arm64 | | Normal
214201679620273 | test-config-link | | Normal
244634136444435 | manual-nonfusion-amd64 | | Normal
9184612610501 | Resource1 | Value1 | Resource
Update a label
Run tw labels update -h to view the required and optional fields for updating labels.
Delete a label
Run tw labels delete -h to view the required and optional fields for deleting labels.
tw labels delete -i 203879852150462
Label '203879852150462' deleted at '97652229034604' workspace
Data links
Run tw data-links -h to view supported data link operations.
Data links allow you to work with public and private cloud storage buckets in Data Explorer in the specified workspace. AWS S3, Azure Blob Storage, and Google Cloud Storage are supported. The full list of operations are:
list: List data links in a workspaceadd: Add a custom data link to a workspaceupdate: Update a custom data link in a workspacedelete: Delete a custom data link from a workspacebrowse: Browse the contents of a data link in a workspace
List data links
Run tw data-links list -h to view all the optional fields for listing data links in a workspace.
If a workspace is not defined, the TOWER_WORKSPACE_ID workspace is used by default. Data links can be one of two types:
v1-cloud-<id>: Cloud data links auto-discovered using credentials attached to the workspace.v1-user-<id>: Custom data links created by users.
tw data-links list -w seqeralabs/showcase
Data links at [seqeralabs / showcase] workspace:
ID | Provider | Name | Resource ref | Region
-------------------------------------------+----------+--------------------------------+-----------------------------------------------------------------+-----------
v1-cloud-833bb845bd9ec1970c4a7b0bb7b8c4ad | aws | e2e-data-explorer-tests-aws | s3://e2e-data-explorer-tests-aws | eu-west-2
v1-cloud-60700a33ec3fae68d424cf948fa8d10c | aws | nf-tower-bucket | s3://nf-tower-bucket | eu-west-1
v1-user-09705781697816b62f9454bc4b9434b4 | aws | vscode-analysis-demo | s3://seqera-development-permanent-bucket/studios-demo/vscode/ | eu-west-2
v1-user-0dede00fabbc4b9e2610261822a2d6ae | aws | seqeralabs-showcase | s3://seqeralabs-showcase | eu-west-1
v1-user-171aa8801cabe4af71500335f193d649 | aws | projectA-rnaseq-analysis | s3://seqeralabs-showcase/demo/nf-core-rnaseq/ | eu-west-1
<snip>
v1-user-bb4fa9625a44721510c47ac1cb97905b | aws | genome-in-a-bottle | s3://giab | us-east-1
v1-user-e7bf26921ba74032bd6ae1870df381fc | aws | NCBI_Sequence_Read_Archive_SRA | s3://sra-pub-src-1/ | us-east-1
Showing from 0 to 99 from a total of 16 entries.
Add a custom data link
Run tw data-links add -h to view all the required and optional fields for adding a custom data link to a workspace.
Users with the workspace MAINTAIN role and above can add custom data links. The data link name, uri, and provider (aws, azure, or google) fields are required. If adding a custom data link for a private bucket, the credentials identifier field is also required. Adding a custom data link for a public bucket doesn't require credentials.
tw data-links add -w seqeralabs/showcase -n FOO -u az://seqeralabs.azure-benchmarking \
-p azure -c seqera_azure_credentials
Data link created:
ID | Provider | Name | Resource ref | Region
------------------------------------------+----------+------+------------------------------------+--------
v1-user-152116183ee325463901430bb9efb8c9 | azure | FOO | az://seqeralabs.azure-benchmarking |
Update a custom data link
Run tw data-links update -h to view all the required and optional fields for updating a custom data link in a workspace. Users with the MAINTAIN role and above for a workspace can update custom data links.
tw data-links update -w seqeralabs/showcase -i v1-user-152116183ee325463901430bb9efb8c9 -n BAR
Data link updated:
ID | Provider | Name | Resource ref | Region
------------------------------------------+----------+------+------------------------------------+--------
v1-user-152116183ee325463901430bb9efb8c9 | azure | BAR | az://seqeralabs.azure-benchmarking |
Delete a custom data link
Run tw data-links delete -h to view all the required and optional fields for deleting a custom data link from a workspace.
Users with the MAINTAIN role and above for a workspace can delete custom data links.
tw data-links delete -w seqeralabs/showcase -i v1-user-152116183ee325463901430bb9efb8c9
Data link 'v1-user-152116183ee325463901430bb9efb8c9' deleted at '138659136604200' workspace.
Browse data link contents
Run tw data-links browse -h to view all the required and optional fields for browsing a data link in a workspace.
Define the data link ID using the required -i or --id argument, which can be found by first using the list operation for a workspace. In the example below, a name is defined to only retrieve data links with names that start with the given word:
tw data-links list -w seqeralabs/showcase -n 1000genomes
Data links at [seqeralabs / showcase] workspace:
ID | Provider | Name | Resource ref | Region
------------------------------------------+----------+-------------+------------------+-----------
v1-user-6d8f44c239e2a098b3e02e918612452a | aws | 1000genomes | s3://1000genomes | us-east-1
Showing from 0 to 99 from a total of 1 entries.
tw data-links browse -w seqeralabs/showcase -i v1-user-6d8f44c239e2a098b3e02e918612452a
Content of 's3://1000genomes' and path 'null':
Type | Name | Size
--------+--------------------------------------------+----------
FILE | 20131219.populations.tsv | 1663
FILE | 20131219.superpopulations.tsv | 97
FILE | CHANGELOG | 257098
FILE | README.alignment_data | 15977
FILE | README.analysis_history | 5289
FILE | README.complete_genomics_data | 5967
FILE | README.crams | 563
FILE | README.ebi_aspera_info | 935
FILE | README.ftp_structure | 8408
FILE | README.pilot_data | 2082
FILE | README.populations | 1938
FILE | README.sequence_data | 7857
FILE | README_missing_files_20150612 | 672
FILE | README_phase3_alignments_sequence_20150526 | 136
FILE | README_phase3_data_move_20150612 | 273
FILE | alignment.index | 3579471
FILE | analysis.sequence.index | 54743580
FILE | exome.alignment.index | 3549051
FILE | sequence.index | 67069489
FOLDER | 1000G_2504_high_coverage/ | 0
FOLDER | alignment_indices/ | 0
FOLDER | changelog_details/ | 0
FOLDER | complete_genomics_indices/ | 0
FOLDER | data/ | 0
FOLDER | hgsv_sv_discovery/ | 0
FOLDER | phase1/ | 0
FOLDER | phase3/ | 0
FOLDER | pilot_data/ | 0
FOLDER | release/ | 0
FOLDER | sequence_indices/ | 0
FOLDER | technical/ | 0
Organizations
Run tw organizations -h to view supported workspace operations.
Organizations are the top-level structure and contain workspaces, members, and teams. You can also add external collaborators to an organization. See Organization management for more information.
Add an organization
Run tw organizations add -h to view the required and optional fields for adding your workspace.
tw organizations add -n TestOrg2 -f 2nd\ Test\ Organization\ LLC -l RSA
Organization 'TestOrg2' with ID '204336622618177' was added
Members
Run tw members -h to view supported member operations.
Manage organization members. Organization membership management requires organization OWNER permissions.
List members
Run tw members list -h view all the optional fields for listing organization members.
tw members list -o TestOrg2
Members for TestOrg2 organization:
ID | Username | Email | Role
-----------------+----------------------+---------------------------------+--------
200954501314303 | user1 | user1@domain.com | MEMBER
277776534946151 | user2 | user2@domain.com | MEMBER
243277166855716 | user3 | user3@domain.com | OWNER
Add a member
Run tw members add -h view all the required and optional fields for adding organization members.
tw members add -u user1@domain.com -o DocTestOrg2
Member 'user1' with ID '134534064600266' was added in organization 'TestOrg2'
Delete a member
Run tw members delete -h view all the required and optional fields for deleting organization members.
tw members delete -u user1 -o TestOrg2
Member 'user1' deleted from organization 'TestOrg2'
Update member role
Run tw members update -h view all the required and optional fields for updating organization members.
tw members update -u user1 -r OWNER -o TestOrg2
Member 'user1' updated to role 'owner' in organization 'TestOrg2'
Leave an organization
Run tw members leave -o <organization_name> to be removed from the given organization's members.
Workspaces
Run tw workspaces -h to view supported workspace operations.
Workspaces provide the context in which a user launches workflow executions, defines the available resources, and manages who can access those resources. Workspaces contain pipelines, runs, actions, datasets, compute environments, credentials, and secrets. Access permissions are controlled with participants, collaborators, and teams.
See User workspaces for more information.
Add a workspace
Workspace management operations require organization OWNER permissions.
Run tw workspaces add -h to view the required and optional fields for adding your workspace.
In the example below, we create a shared workspace to be used for sharing pipelines with other private workspaces. See Shared workspaces for more information.
tw workspaces add --name=shared-workspace --full-name=shared-workspace-for-all --org=my-tower-org --visibility=SHARED
A 'SHARED' workspace 'shared-workspace' added for 'my-tower-org' organization
By default, a workspace is set to private when created.
List workspaces
List all the workspaces in which you are a participant:
tw workspaces list
Workspaces for default user:
Workspace ID | Workspace Name | Organization Name | Organization ID
-----------------+------------------+-------------------+-----------------
26002603030407 | shared-workspace | my-tower-org | 04303000612070
Participants
Run tw participants -h to view supported participant operations.
Manage workspace participants.
The operations listed below require workspace OWNER or ADMIN permissions.
List participants
tw participants list
Participants for 'my-tower-org/shared-workspace' workspace:
ID | Participant Type | Name | Workspace Role
----------------+------------------+-----------------------------+----------------
45678460861822 | MEMBER | user (user@mydomain.com) | owner
Add participants
Run tw participants add -h to view the required and optional fields for adding a participant.
To add a new collaborator to the workspace, use the add subcommand. The default role assigned to a collaborator is Launch.
See Participant roles for more information.
tw participants add --name=collaborator@mydomain.com --type=MEMBER
User 'collaborator' was added as participant to 'shared-workspace' workspace with role 'launch'
Update participant roles
To update the role of a Collaborator to ADMIN or MAINTAIN, use the update subcommand:
tw participants update --name=collaborator@mydomain.com --type=COLLABORATOR --role=MAINTAIN
Participant 'collaborator@mydomain.com' has now role 'maintain' for workspace 'shared-workspace'
Teams
Run tw teams -h to view supported team operations.
Manage organization teams.
Team management operations require organization OWNER permissions.
List teams
Run tw teams list -h to view the required and optional fields for listing teams.
tw teams list -o TestOrg2
Teams for TestOrg2 organization:
Team ID | Team Name | Members Count Name
----------------+-----------+--------------------
84866234211969 | Testing | 1
Add a team
Run tw teams add -h to view the required and optional fields for creating a team.
tw teams add -n team1 -o TestOrg2 -d testing
A 'team1' team added for 'TestOrg2' organization
Delete a team
tw teams delete -i 169283393825479 -o TestOrg2
Team '169283393825479' deleted for TestOrg2 organization
Manage team members
Run tw teams members -h to view the fields and additional commands for managing team members.
To add a new team member, include an existing username or new user email:
tw teams members -t Testing -o TestOrg2 add -m user1@domain.com
Member 'user1' added to team 'Testing' with id '243206491381406'
To delete a team member, include the member's username:
tw teams members -t Testing -o TestOrg2 delete -m user1
Team member 'user1' deleted at 'Testing' team
Collaborators
Run tw collaborators -h view all the required and optional fields for managing organization collaborators.
Manage organization collaborators.
List collaborators
tw collaborators list -o seqeralabs
Collaborators for 88848180287xxx organization:
ID | Username | Email
-----------------+----------------------+--------------------
13136942731xxx | external_user1 | user1@domain.com
127726720173xxx | external_user2 | user2@domain.com
59151157784xxx | external_user3 | user3@domain.com
132868466675xxx | external_user4 | user4@domain.com
178756942629xxx | external_user5 | user5@domain.com
Actions
Run tw actions -h to view supported pipeline action operations.
Actions enable event-based pipeline execution, such as triggering a pipeline launch with a GitHub webhook whenever the pipeline repository is updated.
Add a pipeline action
Run tw actions add -h to view the required and optional fields for adding an action.
Secrets
Run tw secrets -h to view supported workspace secret operations.
Secrets are used to store the keys and tokens used by workflow tasks to interact with external systems, such as a password to connect to an external database or an API token.
Add a workspace secret
Run tw secrets add -h to view the required and optional fields for adding a secret.